BIO 442 MENU
syllabus 
1 - genome
2 - mutate
3 -cell cycle
4 - karyotype
5 - chromoabn
6 -sex-determ
7 -prenatal
8 - mendelian
9 - complex
10 - non-trad
11 - clinical
12 - newborn
13 - teratog
14 - linkage
15 - DNA prof
16 - quanti
17 - links
18 - quizzes
(full title of lecture appears in status bar on the top or at the bottom of your window)

Biology 442 - Human Genetics
Patterns of Inheritance I
As we move into "classical Mendelian genetics" we should first examine some common misconceptions such as genetic = unchangeable; congenital defects are all genetic; all genetic conditions are congenital; genetic conditions show a simple, all or none effect; dominant = prevalent; dominant = all offspring "have it"; skipping of generations (only true for incomplete penetrance and X linked genes); if only males or only females are affected the trait is sex linked. If only one person in the family is affected, the condition is not inherited. Or conversely, if several people in the family have a certain condition, it must be genetic.
Mendelian Inheritance
Traditional Mendelian inheritance refers to the common patterns of inheritance of traits controlled by a single locus (single gene defects) which you learn about in introductory genetics. The general categories are:
1. Autosomal dominant (AD) also codominant and additive dominant
2. Autosomal recessive (AR)
3. X linked dominant (XD)
4. X linked recessive (XR)
5. Y linked or holandric (Y)
(3, 4, and 5 are called sex linked traits)
The terms dominant and recessive refer to the pattern of inheritance of a disorder but reveal something about the gene function. A dominantly inherited disorder generally means the mutant allele is producing a product which changes or interferes with a normal process. These traits are said to be due to gain of function, gain of malfunction or dominant negative mutations....as the case may be. They can also be due to haplo insufficiency when the expression of both alleles is required for normalcy. A recessively inherited disorder/trait would conversely be seen as a "loss of function" situation. A recessive trait is one in which both alleles have to be non functional before the trait or disorder manifests. Another way of looking at it is that one wild-type allele is sufficient to produce enough gene product to cover for a mutant allele. Some dominant traits, however, such as NF1 and retinoblastoma, are due to the inheritance of one (null) mutant allele and one good allele. But the activity of the good gene (you also initially inherited) is knocked out by a somatic mutation or some other event which results in the loss of the heterozygosity (LOH) you enjoyed until then. Codominant inheritance merely means that you can detect the presence of both alleles by their products or activities. This is possible at some level for all genes but we generally mean at the easily observable phenotypic level. Haplo insufficiency is a term given to a disorder due to the loss of activity of one allele. While 50% of the gene product may not be a problem for some cellular processes, this is not always true. This is the situation in the disorder called familial hypercholesterolemia (FH). In FH there are insufficient low density lipoprotein (LDL) receptors in the heterozygote to prevent high levels of cholesterol from building up in the blood vessels. Homozygotes have a much earlier onset due to a an even greater reduction in the number of LDL receptors and, therefore, there is a much greater level of plasma LDL cholesterol. So while FH is said to be dominantly inherited, a double dose in the homozygote results in an early lethal.
You have learned and will learn more about genetic traits that are non-traditional such as trinucleotide repeats (TNRs), microdeletion syndromes, mitochondrial mutations, uniparental disomy, etc. The inheritance of these may not fit neatly into the traditional inheritance patterns. Nor may the causes be neatly classified into our mutation categories. Except for Friedreich Ataxia, the TNRs are AD but the expansion which causes them is not a "neat" point mutation. The microdeletion syndromes are AD in that only one chromosome need have the deletion for the disorder to be expressed but the mutation involves many genes being deleted not just a few nucleotides. The mitochondrial mutations are point mutations but the inheritance pattern is complex due to heteroplasmy and sometimes the interactions with nuclear gene products. The problems that arise due to uniparental disomy are due to an "epigenetic" effect. The genes which are "imprinted" are not permanently changed and the imprinting pattern will be reversed in the gametes of the affected individual.
Autosomal Dominant
We will begin with autosomal dominant (AD). The genes for these traits are on the autosomes. Dominant does not mean prevalent, dominant traits can be rare (achondroplasia, TNR, NF1, etc.). Dominant means you need only one mutant gene to express the trait. In general, the heterozygote and homozygote for the mutation show the same phenotype. However, since some AD traits are rare, one may not have an opportunity to see the homozygous phenotype. For example, achondroplasia in a double dose is lethal. The phenotype is very similar to a related skeletal lethal called thanatophoric dwarfism. The mutations for achondroplasia and thanatophoric dwarfism are in related growth factor receptor genes (we will discuss more later). The lethality of the homozygous condition was not appreciated until achondroplastic persons married and it was noted that approximately 25% of the fetuses from such unions did not survive. Therefore, achondroplasia is not a "true" dominant, it is an "additive dominant" since it shows a different or intermediate phenotype when there is only one mutant allele as opposed to two in the homozygote. On the other hand, Huntington disease is a true dominant. Homozygotes are known and they do not have a more severe phenotype. Other rare dominants may prove to be additive. Familial hypercholesterolemia, mentioned earlier, is another example of an additive dominant. In FH the homozygote is affected earlier than the heterozygote for the mutant alleles.
Autosomal dominant traits are often associated with malformations (chondrodystrophies and the FGFR genes) or other physical features (Treacher Collins); show pleiotropy (Marfan with mutant fibrillin gene); are clinically variable (NF1); are less severe than recessive disorders; are age dependent (may manifest later in life); are usually due to the presence of an abnormal protein (gain of function) but some are due to haploinsufficiency (loss of function). AR disorders such as those involving enzymes are generally loss of function situations.
Some dominant traits (NF1 and retinoblastoma) are due to the loss of heterozygosity which can occur by a variety of mechanisms including somatic mutation, loss of a chromosome, mitotic recombination, etc.
Autosomal dominant traits are often complicated by variable expressivity (VE) and incomplete penetrance (IP).
Variable expressivity refers to quantitative and qualitative differences in phenotype between individuals having the same allele or genotype. It refers to the fact that not all members of a family may show all possible features of the phenotype. The severity, frequency of "attacks," age of onset all can vary. The causes of variability include the other genes, possible imprinting, the sex, maternal factors such as cytoplasmic inheritance, and pre and post natal environment, nutrition, medications, etc. If the trait is X linked, there can be variation due to the differences in the pattern of X inactivation in heterozygotes. VE refers to the fact that not everyone affected, even in the same family, who are assumed to have the same mutation, has exactly the same phenotype. Some traits such as Treacher Collins are fully penetrant (all carriers are affected) but highly variable in expression (each individual may express it differently even in the same family). The cause of intra familial variability, where all affected are assumed to have the same gene, is not known but it is thought to be due to other unrelated genes that vary for each family member which influence the expression of the gene responsible for the trait studied.
And some traits may show incomplete penetrance (IP) where the mutation may not be expressed in all carriers/heterozygotes. Penetrance is an "all or none" phenomenon and refers to the fact that not everyone with the same allele or genotype expresses it phenotypically. The term is used primarily in reference to dominant traits. The causes of incomplete penetrance are the same as those given for variable expressivity. If a condition is expressed in less than 100% of persons known to carry the allele (obligate heterozygotes), the trait is said to show reduced penetrance and the percent can be calculated for each specific disorder. Obligate heterozygotes are those individuals in the direct line of descent of an affected parent who have affected children. The percent penetrance of a trait is calculated by determining the number of obligate heterozygotes with affected children that are and are not affected themselves. The fraction of those expressing the trait over the total number of those with affected children will give you the percent penetrance. For example, polydactyly can express with extra digits (one or more) on one or both hands, and/or one or both feet (VE). It can be inherited from a parent who has an affected parent and sibs but who does not have any extra digits (IP). For any one trait a frequency of penetrance can be determined empirically by counting, in a large number of pedigrees, the number of unaffected obligate carriers (with affected children) and the number of affected carriers who have affected children. So the percent penetrance is based on pedigrees and does not refer to expressivity in a single individual. The percent of penetrance is sometimes used in genetic counseling to inform an individual with an affected parent and sibs, that while they are unaffected there is the probability that they are carriers.
Susceptibility genes for common, complex disorders such as breast cancer, Alzheimer Disease, alcoholism and asthma, are often inherited in an AD pattern. They are often sex influenced and show variable expressivity and incomplete penetrance.
The AD trait, Marfan syndrome and homocystinuria (AR) are examples of locus heterogeneity in that they have similar phenotypes (dislocated lens, long extremities) but Marfan is due to a mutant structural protein, fibrillin, and homocystinuria is an enzyme defect due to cystathionine synthase deficiency (which happens to be vitamin responsive). The definition of locus heterogeneity = same (or similar) phenotype but different mutant genes (at different loci). Examples are XP and mucopolysaccharidoses (we will study later). If we consider ambiguous genitalia a phenotype then 5 reductase deficiency and CAH could be considered examples of locus heterogeneity. It is important for genetic counselors when providing recurrence risks, to be aware of the different modes of inheritance of traits with similar clinical findings.
Sporadic, new mutations, are usually autosomal dominants (they can also be XR or XD). (New autosomal recessive mutations occur as frequently but are not observed since it takes two!) On average, the age of fathers of sporadic cases is advanced (paternal age effect) due to increasing risk of new mutations. The frequency of sporadic cases (new mutations) is positively correlated with the severity of the phenotype. In other words, the greater the reproductive fitness of affected individuals, the less likely any given case resulted from a new mutation. For example, Progeria or premature aging, is a genetic lethal and is only due to new mutations. Sibs have been affected but it is due to gonadal mosaicism. Cornelia de Lange Syndrome and Incontinentia Pigmenti
The characteristics of autosomal dominant inheritance are (usually)
a. Vertical pattern in a pedigree (multiple generations affected). The phenotype appears in every generation except when cases originate by new mutations in a phenotypically unaffected parent or when the disorder is non penetrant or is expressed very mildly. With these exceptions, unaffected family members will not transmit the trait.
b. Males and females affected equally frequently and severely. Exceptions are sex limited or sex influenced dominant traits.
c. If the trait is rare, each affected person is heterozygous; s/he inherits the gene from only one parent.
d. When an affected person mates with an unaffected person, each offspring has a 50% chance of inheriting the affected phenotype regardless of the sex of the affected parent. This reflects the fact that for rare AD traits each affected person is heterozygous.
e. Male to male transmission occurs.
f. Variable expressivity and Incomplete penetrance are common in AD traits.
g. New AD sporadics occurs and are often correlated with paternal age.
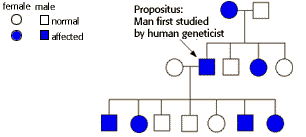
Autosomal Dominant Pedigree
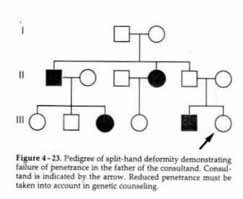
Autosomal Dominant with Incomplete Penetrance
Sex Limited Autosomal Dominant
Autosomal Recessive
Autosomal recessive disorders are those which require the affected person to have two mutant alleles. The parents of a person with AR disorder are obligate heterozygotes. They are usually not affected. Their probability of having another affected child is 25%. The normal sibs of an affected child have a 2/3 probability of being a carrier (draw your Punnett square and see!). Both males and females are affected with equal severity unless the trait is sex influenced or sex limited. If two people with the same AR disorder have children, all of their children will be affected. AR traits are often more severe than AD traits and AR phenotypes are less variable than AD phenotypes.
If someone has a rare AR disorder (which means there are not a lot of heterozygotes around) then s/he may be the result of a consanguineous mating or the parents may be from a small village where people are more apt to be related to one another. The rarer the recessive phenotype, the more likely the parents are to be consanguineous.
Some AR traits are what are loosely referred to as "ethnic diseases." What is meant is that the gene is more prevalent in that ethnic group. Examples are sickle cell disease in African Americans, thalassemia in Mediterranean people, Tay Sachs and Gaucher disease in Ashkenasi Jews and cystic fibrosis in Northern Europeans. Often AR pedigrees can be traced back to a common ancestral couple in which the mutation first occurred. This is called a Founder Effect.
Sickle cell disease shows an AR pattern of inheritance. However, sickle cell trait which is the name given to the carrier status is "co-dominant" since the mutant allele (HbS) produces mutant hemoglobin and the normal allele (HbA) produces normal hemoglobin. In the case of sickle cell we know that the high incidence of carriers among African Americans has to do with heterozygote selection. The carriers (heterozygotes), who are not affected with the disorder, were protected from malaria because the malarial parasite did not like their mutant hemoglobin and left them alone. Homozygous HbAA died from malaria and homozygous HbSS died from sickle cell disease. Therefore, the gene in the heterozygous state was selected for in the populations where malaria was rampant. The same situation is true for the thalassemia heterozygotes who were also protected from malaria in the Mediterranean area. thalassemia is also caused by mutations in the globin gene. Although the basis of the higher incidence of other ethnic disorders is less clear, it is assumed that heterozygotes may have had a yet undiscovered advantage in those populations (people are working on this question.)
In most families where an AR disorder appears, it may be the first and only case in the family although sibs are affected. If the trait is relatively common or if there is consanguinity in the family, more than one generation can be affected. Both males and females are affected and both can pass the trait on. This is not true if the disorder or some aspect of the phenotype it is sex limited. Examples are 5 reductase deficiency which only manifests itself in males and CAH which only causes ambiguous genitalia in females. However, CAH often has a "salt wasting" component to the phenotype which is equally expressed in both males and females.
Enzyme deficiencies are all AR with the one exception, Acute Intermittent Porphyria (AIP). Inborn errors of metabolism is a term often associated with enzyme deficiency disorders. Heterozygote show a definite dosage effect and usually produce approximately 50% of the normal amount of the enzyme. Some mutations cause no gene product to be made, some cause less gene product to be made and some make a nonfunctional gene product. Fifty percent of the enzyme is sufficient to produce a normal phenotype although carrier detection and prenatal diagnosis may rely on picking up those who have less than the normal amount of the enzyme activity. Some mutations involved in AR enzyme disorders can affect the production of the enzyme by interfering with transcription or translation thereby producing no product from the mutant allele (known as null alleles). Mutations can also affect the active site whereby the substrate, coenzyme or product are held more loosely or more tightly or not at all; these are often referred to as Km mutations. Mutations in an allosteric site can change the regulation of the enzyme activity by affecting the binding of regulatory molecules. Mutations which result in a protein that is similar in quaternary structure to the normal gene protein are called CRM+ mutants because they cross react with antibodies made to the normal protein. CRM means "cross reacting material." A mutant gene either makes a CRM+ or CRM- product. This means that the protein product either does (+) or does not (-) react to the antibodies made to the normal gene product. CRM- proteins are those that do not retain a resemblance to the normal gene protein. Sometimes CRM+ mutants can be corrected by adding more substrate or coenzyme to increase the reaction product. Individuals with CRM+ mutants may also be able to use enzyme replacement therapy without making antibodies to the enzyme.
As a consequence of allelic heterogeneity many individuals with any particular AR disorder are compound heterozygotes. The consequence of having several possible mutant alleles in the population for any gene is that not all individuals with mutations in the same gene will have the same phenotype. This results in inter familial differences. This situation is different from variable expressivity where we are talking about intra familial difference of expression where it is assumed all members of the family share the exact same genotype.
The presence of multiple alleles (FH has 400) for disease genes complicates DNA testing and diagnosis since not all mutations can always be identified. While gene sequencing is becoming more common, there is still a problem due to commonly finding normal polymorphisms (SNPs, single nucleotide polymorphisms) which may cause no problem. Sometimes molecular geneticists can guess whether these SNPs could cause a problem by what amnio acid is changed and where it is in the protein product. It is important to be aware that the pedigrees of families with AR traits may appear to be AD because of consanguinity and/or the high frequency of carriers of the gene for the recessive trait. For example, if a person with an AR form of albinism marries his/her cousin who is a carrier then half of their children (both boys and girls) will be albino. This is known as quasi or pseudo dominance.
All humans are heterozygous for recessive alleles that, if present homozygously, would be lethal. This is sometimes referred to as the "genetic load."
The characteristics of traits with an autosomal recessive inheritance pattern (usually)
1. Show a "horizontal" pattern (as opposed to vertical seen in AD). This means that if the condition is rare, siblings may be affected but rarely are their parents or children.
2. Both sexes can be affected. 3. The parents of an affected child are both carriers but themselves unaffected.
4. If the trait is rare, one may suspect consanguinity.
5. The recurrence risk for each sib of an affected person is 1/4 (25%).
6. The probability of a normal sib being a carrier is 2/3.
Sex linked traits vs. sex influenced and sex limited traits
Sex linked refers to all genes on the X or Y (non pseudoautosomal region). Linked is a reference to the fact that all genes on any one chromosome are said to be "linked." Most sex linked genes are X-linked genes because of the paucity of gene on the Y. Most of these genes have nothing to do with the determination of sex. Sex influenced and sex limited traits are usually coded for by genes on the autosomes. Their expression is influenced by the sex of the individual. Sex limited genes are not expressed (not penetrant) in one of the two sexes. The traits include all sex specific secondary sexual characteristics (breasts, muscle mass, hair and fat distribution) and the genitalia. Sex influenced traits are found in both sexes but are expressed differently or in a different inheritance pattern. For example, baldness is dominant in males and recessive in females and height (a multifactorial trait) is influenced by the sex of the individual. Hormones and other genes affect the expression of sex limited and sex influenced traits. Castrated men do not get bald.
Sex linked inheritance refers to traits controlled by genes on the X or Y chromosome. There are more genes on the X than the Y so we will start with X-linked inheritance. When analyzing sex linked traits it is a good idea to use the sex chromosome symbols and attach the genes as appropriate. For example, XD Xd and XD Y, etc. Males are said to be hemizygous for X linked traits. It is important to note that whether a female who is heterozygous for an X linked gene is counted as affected and whether the phenotype is called XR or XD depends often on the sensitivity of the clinical test or evaluation. Whether a female carrier of an X linked recessive is affected depends on whether there is skewed X inactivation.
X linked dominant
When the disorder is nearly always manifest in heterozygous females it is referred to as X-linked dominant (XD). Hypophosphatemic rickets, a.k.a. Vitamin D resistant rickets, is such a trait. Females tend to be affected twice as often as males and an affected female will transmit the phenotype to 50% of her children independent of their sex. All the daughters of an affected male will be affected but none of his sons will be affected.
An X-linked prenatal male lethal is also an example of XD. Incontinentia Pigmenti and Rett Syndrome are examples of such traits. Only females are affected and they will pass the disorder to 50% of their daughters. Because it is a male fetal lethal, affected females will have fewer sons than daughters and will show an increased frequency of spontaneous abortion (SAB), these losses are hemizygous male fetuses.
In general, XD traits that are not lethal, are more mildly expressed in the female than in the male. The Coffin-Lowry Syndrome is a good example. The hemizygous males are severely affected. They are mentally retarded, short stature, coarse features, malocclusion, tapered fingers, scoliosis and other defects while the carrier females have slight to moderate mental deficiency, mild facial changes, tapered fingers and short stature and some may be completely normal.
Twice as many females are affected with an XD trait since they have twice as many X chromosomes. Rare XD alleles will be found in hemizygous males and heterozygous females. As for the XR traits, the frequency of the XD mutant allele will be equal to the number of affected males. However, the frequency of affected females will be 2pq since they are heterozygous. Hence the frequency of affected females will be 2 x 1 x q (p is more common and very close to 1) thus twice as frequent as affected males.
Summary of Characteristics of X Linked Dominant Disorders
1. All daughters of a male affected with a rare XD are affected and no sons are affected.
2. When a female is affected with a rare XD, the pedigree is the same as for AD: 50% of both male and female children are affected.
3. For rare XD traits, twice as many females are affected as males.
X linked recessive
When a male has a mutant X-linked recessive allele he will express the trait since he is hemizygous. He has no other X to cover for him as the female does or as all autosomal mutant alleles have. Males are usually more severely affected than females.
XR traits show no male to male transmission since their sons receive their Y and not their X. Obviously, unaffected males do not transmit the phenotype. All daughters of an affected male are heterozygous carriers since they get their father's X chromosome with the mutant allele. Female heterozygotes (carriers) may express the trait due to "unfortunate Lyonization."
Some mothers of affected males are not carriers but instead the new mutation has arisen on the X-linked allele of the mother. When the disorder is a genetic lethal in males (affected males do not reproduce) about 2/3 of affected males have a carrier mother and the other 1/3 arise by new mutation in the mother. An example of this is Duchenne muscular dystrophy. A carrier mother who appears to be the first one in her family, often has a father who was of advanced age when she was born. In other words, her carrier status is the result of a new mutation in her father. Her son will be the first to show the trait since the mutation does not (usually) manifest in a carrier.
X-linked phenotypes are often clinically variable, particularly in heterozygous females due to differences in X inactivation. Sometimes they are suspected of being autosomal dominant with non penetrance. AI (androgen insensitivity) was thought to be AD with sex limited expression until the gene was identified.
Hemophilia is genetically heterogeneous but the most common type, hemophilia A, is XR and due to a deficiency of factor VIII a blood protein needed for normal fibrin formation for clotting. Queen Victoria was a carrier and many of her progeny were either carrier females or affected males.
If a male with an X linked recessive trait has a child with a heterozygous female, 50% of the sons will be affected thus giving the false impression of male to male transmission. 50% of the daughters of these matings will be as severely affected as their hemizygous brothers. In a small pedigree, this pattern may look like AD inheritance (pseudo or quasi dominant).
Although inbreeding can have consequences due to the greater probability of related persons carrying the same recessive lethal or semi lethal genes for AR traits, this is not true for XR traits. Consanguinity in a family with an XR such as hemophilia could result in affected females, however.
Summary of Characteristics of X Linked Recessive Disorders
1. For rare traits, more males are affected than females (square root of the frequency of affected females).
2. Female carriers are not expected to be affected unless there is skewed X inactivation.
3. The daughters of an affected man will all be carriers. So the a priori risk for his grand sons to be affected is 50%.
4. The sons of an affected male will never inherit the gene.
5. In families with males affected with a rare XR, the males will all be related through the females of the family.
6. A significant number of isolated cases are due to new mutations which are correlated with grandpaternal age.
For X linked disorders, the frequency of the mutant phenotype in males is equal to q. In other words, the gene frequency is equal to the "people frequency" for these XL traits. The frequency of an XR trait in females is then q2. The frequency of XR carrier females is twice the frequency of affected males (2pq) since p is very close to one.
If the trait such as Duchenne Muscular Dystrophy (DMD) is a genetic lethal, then the males do not survive long enough to reproduce and thus no females will be affected. This is because affected females would have to have inherited the mutant gene from each parent. When a case of an XR disorder occurs in a family with no other affected individuals, the new mutation may have arisen maternal grandfather. This is related to the age of the grandfather and is a "grand paternal age" effect. X linked disorders may manifest in females due to unfortunate lyonization where the X chromosome with the normal allele is inactivated in significantly more than 50% of the woman's cells.
A COMPARISON OF THE MAJOR ATTRIBUTES OF XD AND XR PATTERNS OF INHERITANCE
|
XD
|
XR
|
|
| Recurrence risk for heterozygous female x normal male matings |
50% of sons affected |
50% of sons affected |
| Recurrence risk for affected male x normal female matings |
0% sons affected |
0% sons affected |
| Transmission pattern |
Vertical transmission pattern |
Generation skipping may be seen because of transmission through carrier females |
| Sex ratio | 2x as many males affected as females, unless it is a male lethal | The proportion of affected females is the square of the proportion of affected males (e.g., when 1 in 10 males are affected then 1 in 100 females are affected) |
| Other |
No male to male transmission |
No male to male transmission |
Y linked inheritance
Y linked inheritance is called "holandric" inheritance. The Y-linked genes are on the non pseudoautosomal region of the Y. Only males have these genes and any mutations would result in all sons of an affected male being affected and none of his daughters.
The X chromosome is large, it belongs to the C group chromosomes and contains 6% of the total DNA. The Y chromosome, on the other hand, is small and belongs to the G group chromosomes. 250 or more disorders have been mapped to the X chromosome but only 20 to the Y. As mentioned above, traits coded for by genes on the Y chromosome are said to be holandric. Known genes on the Y include SRY, genes influencing height, genes for tooth size and those controlling spermatogenesis. There are regions of the Y which are homologous with the X but often the genes are non functional (pseudogenes). There is evidence that the X gave birth to the Y. The Y chromosome is not essential for viability.
The identification of Thomas Jefferson as the father of at least one son of his slave, Sally Hemings, was done using Y linked markers (holandric). Since Jefferson had no sons by his wife but he did have brothers, the Y chromosomes studied were from the descendants of his paternal grandfather. Most of the Y chromosome is passed intact from father to son, so it can be used to trace paternal lineages. However, such studies require enough polymorphic markers (small regions of DNA that vary among individuals) so that Y chromosomes can be distinguished by the haplotype that they carry. (Haplotype refers to a series of tightly linked genes inherited as a unit. Each of the gene loci have multiple (polymorphic) alleles.)
Researchers from several laboratories have identified a collection of suitable markers from the Y chromosome over the past two years, and this collection is now being used in male-line genetic studies. Foster, E.A. et al Nature 396, 27-28 (1998) examined a haplotype containing 19 polymorphic markers. Jefferson's haplotype (inferred from male-line descendants of his paternal grandfather) seems to be quite rare, inasmuch as it was not seen among a sample of 670 Europeans or 1200 people worldwide. The authors found that this rare haplotype perfectly matches that of Sally's son, Eston Hemings, male-line descendants. The probability of such a match arising by chance is low...safely less than 1%. Together with the circumstantial evidence, it seems to seal the case that Jefferson was the father of Eston Hemings. Jefferson's haplotype does not match male descendants of Sally's first son, Tom Woodson, however. The simplest explanation is that Jefferson was not Tom's father. An alternative explanation would require non-paternities among Tom's offspring. The jury remains out with respect to Sally's other children, but the burden of proof has clearly shifted.
The Cohains, a Jewish sect, trace their lineage back to the biblical Aaron. A study of their Y chromosomes showed 90% identity among the Cohains.
Genetic Drift and Founder Effects
Mutant alleles which arise or became common in a specific population due to inbreeding [e.g., Ellis van Creveld dwarfism (AR) in the Amish], or which are introduced into a specific inbred population [e.g., HD (AD) in Venezuela], or which are selected for in specific populations [e.g., HbS in Africans; thalassemia in the Mediterranean (both AR)] may be more prevalent in that same population or ethnic group either due to genetic drift or heterozygote selection. If the rare gene was introduced into a group and the group is isolated or inbred, this is referred to as the founder effect. This also is seen on a larger scale when there is a higher incidence of a particular mutant allele in an ethnic group. For example, there are many mutant genes that cause cystic fibrosis but the most common one is the delta F 508 mutation and the carrier frequency of this allele is highest among descendants of northern Europeans. The frequency of each of the different mutant alleles for CF vary by the ethnic group. The persistence of certain deleterious alleles in specific populations may be due to selection for the deleterious gene in the carrier. The best documented example of this is the HbS or sickle cell allele. This allele in the homozygous state resulted in death until recently. The gene in the heterozygous state (sickle cell trait) was protective against the parasite that causes malaria and thus the allele persisted where malaria occurred. It takes a loooooooooong time for a deleterious allele to disappear..
From the incidence of a rare AR disorder in a population, one can calculate the frequency of carriers (heterozygotes) by using the Hardy Weinberg equation. If p = the frequency of
the normal allele and q = the frequency of the mutant alleles then p2 = the frequency of homozygous normal individuals and 2pq = the frequency of heterozygotes (carriers) and q2 = the frequency of individuals affected with the disorder. If you know the incidence of the disorder, you can determine the carrier frequency. Since the gene frequency, q, is very small for a rare trait, the value of p is approximately equal to one. If the incidence of sickle cell disease is 1/400 African Americans (q2), the gene frequency (q) of the mutant allele is 1/20. The carrier frequency is then 2 x 1 x 1/20 or 1/10. (Do not confuse people frequencies and gene frequencies.)
Gene frequencies for codominant traits such as the inheritance of microsatellites (STRs) can be calculated directly by counting the number of each allele and dividing by the total number of genes. (In our class exercise we each reported the number of repeats we had in one pair of alleles where the variation in size was between 1 and 5 repeats.) Because of the large number of different size repeats in a population, these "markers" are useful in population studies, DNA fingerprinting, and paternity testing. The frequency of these (STR repeat) alleles, like any other, also vary by populations or ethnic groups and thus the laboratories that use them keep big data banks of allelic frequencies for each of the ethnic groups with which they work.
