BIO 442 MENU
syllabus 
1 - genome
2 - mutate
3 -cell cycle
4 - karyotype
5 - chromoabn
6 -sex-determ
7 -prenatal
8 - mendelian
9 - complex
10 - non-trad
11 - clinical
12 - newborn
13 - teratog
14 - linkage
15 - DNA prof
16 - quanti
17 - links
18 - quizzes
(full title of lecture appears in status bar on the top or at the bottom of your window)

Biology 442 - Human Genetics
Non Traditional Inheritance
Microdeletions or Contiguous Gene Syndromes
Microdeletion syndromes are also known as contiguous gene syndromes and segmental aneusomy. They cannot always be detected by ordinary karyotyping techniques so detection requires high resolution banding (HRB) or FISH probes. Many laboratories are now offering a microdeletion panel. Many genes are involved in these complex but recognizable phenotypes. Some of the more common microdeletion syndromes are William syndrome, DiGeorge (a.k.a. velocardiofacial and Shprintzen), Miller-Dieker lissencephaly, Prader Willi and Angelman, retinoblastoma. The latter three syndromes can also be caused by mutations in some of the key genes in the duplication area. Read about these in your textbook. Microdeletions and microduplications involve many genes but are usually not detectible by ordinary Giemsa banding. High resolution (prometaphase) banding can pick up some but if you suspect on the basis of a clinical diagnosis that a person has a microdeletion syndrome, you can request the cytogenetic lab do FISH for the region in question. Do not be misled by the "micro" in microdeletion. Many genes are deleted. Microdeletions are contrasted to "macrodeletions" such as 4p- (Wolff-Hirschhorn) and 5p- (Cri du chat) which involve even greater amounts of chromosome and, therefore, gene loss.
Subtelomeric rearrangements are often found in babies with low birth weight and microcephaly. GTG banding is always done followed by subtelomeric FISH probes. If a deletion is found in the child, a balanced chromosome rearrangement involving the same region may be found in one of the parents' chromosomes. Telomeric deletions have been found in 4-7% of idiopathic mental retardation. Microarrays of BAC clones for each chromosome are being developed to detect deletions and additions of chromosome material. )
Microdeletion Syndromes diagnosable with FISH
| Syndrome | Chromosome Region | Probe target |
| Wolff-Hirschhorn syndrome | 4p16.3 | Wolff-Hirschhorn critical region |
| Cri du Chat | 5p15 | |
| Williams-Beuren syndrome | 7q11.23 | Elastin gene |
| Retinoblastoma | 13q14 | |
| Prader-Willi/ Angelman syndrome | 15q11-q13 | PW/AS critical region |
| Smith-Magenis syndrome | 17p11.2 | Smith-Magenis critical region |
| Miller-Dieker syndrome | 17p13.3 | Lissencephaly gene |
| Rubinstein-Taybi syndrome | 16p13.3 | |
| DiGeorge/Velo-Cardio-Facial syndrome | 22q11.2 | DiGeorge/VCFS critical region |
| Kallman syndrome | Xp22.3 | KAL gene |
| Steroid sulfatase deficiency (ichthyosis) | Xp22.3 | Steroid sulfatase gene |
Chromosomes from various sources including amniotic fluid, chorionic villi, peripheral blood, and tissue samples can be used for FISH to rule out microdeletion syndromes. Peripheral blood specimens - 5-10 ml of blood in a sodium heparin vacutainer (green top); Amniotic fluid specimens - 15 ml of amniotic fluid; Chorionic villi samples - 10-20 mg of chorionic villi. FISH should be ordered in conjunction with chromosomes with the following exceptions: chromosomes have previously been done; there is a family history of a microdeletion; the diagnosis is firm and no other syndromes are included in the differential diagnosis.
Both microdeletions and microduplications are inherited in an AD pattern. After the birth of a child with one of these syndromes, parents should be counseled about monitoring of subsequent pregnancies by "FISHing" because of possible gonadal mosaicism in one parent or cryptic parental translocations.
Retinoblastoma is a relatively common childhood cancer of the eye. It is due to a mutation or a deletion at 13q14. The deletion may be visible by G banding or HRB or it may require FISH. The gene whose function is lost in this cancer is a "tumor suppressor gene" (as is the BRCA1 and BRCA2 gene for breast cancer). One deletion or mutation is inherited from a parent and then a somatic mutation causes the "good" allele to be mutated or deleted. Cancer is due to somatic mutations often in DNA repair systems and cell cycle control genes. Usually several mutations are needed to wipe out a normal cell cycle control mechanism. When a person inherits one mutation in a cell cycle control gene, another somatic mutation (or deletion) is likely to occur during their lifetime. The result is the "loss of heterozygosity" of that locus....the one good gene was able to maintain cell division control but with both gone, the control is lost and cancer results. However, unlike retinoblastoma, most cancers do not develop until several cell cycle control genes are knocked out. Familial or inherited cancer is when a person inherits one mutation in a critical gene. The somatic mutation occurs later. One suspects inherited cancer when the cancer is bilateral (e.g., breast, kidney) and has an early onset (before 40 years).
DiGeorge and Velocardiofacial (Shprintzen) syndrome can be picked up with the same FISH probe at 22q11.2; Prader-Willi syndrome (PWS) and Angelman syndrome (AS) can also be picked up by the same probe for 15q11.2. The size of the deletion in AS is larger than for PWS and is distal to it. Probes for Miller-Dieker lissencephaly (smooth brain) are 17p13.3 and for Smith Magenis at 17p11.2. These are too far apart on 17 p to be picked up by the same probe. Isolated lissencephaly occurs when a smaller region including the L1S1 gene at 17p13.3 is deleted or mutated. The L1S1 gene and a homologous gene on chromosome 2 code for G proteins probably involved in signal transduction crucial for cerebral development. The critical gene responsible for Smith-Magenis is proximal to the CMT 1A gene (see below), it codes for a small nuclear RNA U3.
Williams is due to a deletion at 7q11.23 and the deletions include the elastin gene. It can arise from a cryptic parental translocation, especially in the telomeric region. Williams syndrome patients have an "elfin" appearance and a loquacious "cocktail party" personality. Williams syndrome because of its unique behavioral phenotype and the velo-cardio-facial syndrome because of its association with bipolar disorder and schizophrenia have proven useful to behavioral and psychiatric geneticists who are trying to locate genes controlling mental illness and behavior.
Microduplications can also be picked up by FISH probes. Beckwith Wiedeman, a congenital disorder which results in macroglossia, macrosomia, organomegaly and omphalocoele, is due to a duplication of 11p15. It arises from an unbalanced translocation possibly from a parental balanced translocation or recombinant chromosome from parental pericentric inversion. Charcot-Marie-Tooth syndrome (CMT 1A), an adult onset peripheral neuropathy, can be caused by a gene duplication at 17p11.2. The FISH studies are done on interphase nuclei (instead of metaphase chromosomes) since the duplication is small and the two signals can be seen only on elongated chromatin. The duplication form may be due to flanking repeats (which cause mispairing) found nearby. Alu repeats have been found at the boundaries of both duplications and deletions.
Some Cri-du-Chat patients may have a deletion of the entire 5p, however, the critical region for the clinical phenotype involves the critical region 5p15.2. The high pitched cry maps to 5p15.3 while the remaining features map to a small region of 5p15.2. It is estimated that about 100 genes reside in this region. Deletions that do not include these 5p15.2/15.3 present varying clinical phenotypes from severe mental retardation and microcephaly to clinically normal. 90-85% are de novo, 10-15% are due to unequal segregation of parental translocations, and 0.3% are due to parental mosaicism. FISH is used for Cri-du-Chat and Wolff-Hirschhorn (4p-) if the clinical diagnosis is not confirmed up by ordinary G banding or HRB. The critical region for 4p- is quite small and cannot always be seen on HRB.
The fact that the same probe picks up two different microdeletion syndromes does not mean that the same gene losses are involved in those syndromes. In fact, the candidate gene for Prader-Willi is the SNRPN gene which codes for a snRNP (small nuclear ribonucleoprotein N) particle used in RNA splicing. The candidate gene for Angelman is UBE3A, a gene that codes for a ubiquitin protein ligase, which transfers ubiquitin to protein substrates (for proteolysis).
Genomic Imprinting (Parent of Origin Effects)
and Uniparental Disomy (Heterodisomy and Isodisomy)
A child's genes are not all equal. In some cases, the copy from either the mother or the father is turned off. This affects the child's ability to acquire resources in the womb, after birth, and perhaps throughout life. Genomic imprinting involves the epigenetic inactivation of one allele of a gene, resulting in monoallelic expression that is parent-of-origin dependent.
Examples of imprinting are Mules and Hinneys; Moles (2n paternal only); Teratomas (2n maternal only). We now understand that both maternal and paternal contributions are required for normal development. The paternal contribution is important for extra embryonic development. The maternal contribution isimportant for fetal development. Imprinted genes influence transfer of nutrients to the fetus and the newborn from the mother and affect growth in the womb and behavior after birth.
KNOWN IMPRINTED REGIONS OF CHROMOSOMES
Imprinting is a phenomenon that selectively silences only certain genes. Different genes are silenced in spermatogenesis than in oögenesis. Imprinting is referred to as an epigenetic phenomenon meaning that the effect on the genes is not permanent. It involves reversible transcriptional inactivation resulting in monosomy for the gene(s) involved. DNA methylation, is the key molecular mechanism. This is similar to the X-inactivation mechanisn, another epigenetic phenomenon. Genome wide demethylation (erasure) occurs in germ cells and remethylation follows (resetting). The process may continue into the early stages of embryonic development as evidenced by the higher than expected incidence of imprinting disorders among offspring from IVF (in vitro fertilization.
REVERSING AND RENEWAL OF IMPRINTING
What we learn from Prader Willis and Angelman syndromes, from hydatiform moles and teratomas and from mules and hinnies, is that the genetic contributions of mammalian fathers and mothers are not the same. However, the imprinting is reversible and what was imprinted in the paternal contribution will become un imprinted in his female children and what was imprinted in the maternal contribution will become unimprinted in her male offspring.
IMPRINTING AND DISEASE
Prader-Willi syndrome is characterized by lack of normal amount of fetal movement, hypotonia, genital hypoplasia, mental retardation, short stature, small hands and feet, almond eyes, hyperphagia leading to obesity. 70-80% are due to paternal deletions detectible by HRB or FISH. 20-30% are due to maternal uniparental disomy (UPD) usually heterodisomy (two different maternally derived chromosomes 15) caused by maternal non disjunction in Meiosis I. This is associated with maternal age. Occasionally, there is isodisomy (two copies of the same maternal chromosome 15) due to non disjunction in Meiosis II. Isodisomy has additional problems since the person in homozygous for all the alleles on 15 and may result in an AR disorder since we all carry recessive deleterious genes for which we are usually heterozygous. The syndrome can also be caused by a point mutation in the paternal gene or mutations in the genes controlling the imprinting process (methylation).
Uniparental Disomy (UPD) can be detected by DNA polymorphic markers in the region or by use of a methylation sensitive enzyme and a specific probe. The latter tests are expensive and only done by very few diagnostic labs so if a deletion is not found, the diagnosis is made clinically based on the phenotype. The syndrome is due to the loss of the paternal allele. The maternal allele is turned off (imprinted) by methylation during meiosis and is not functional. The paternal allele can be lost due to a deletion, non functional due to a mutation or absent because both chromosomes 15 are of maternal origin (UPD). UCLA has a Prader-Willi Clinic.
Angelman is also called the "Happy Puppet" syndrome. It is characterized by mental retardation, global developmental delay, ataxia, inappropriate laughter, seizures, microcephaly, hand flapping. Angelman is due to the loss of an active maternal allele. This loss can be by deletion (70%), familial point mutation (30%) or, rarely, paternal uniparental disomy. In AS, the UPD is usually due to a non disjunction in paternal Meiosis II or in mitosis. In either event, two copies of the paternal 15 are present and none of the maternal 15. To be normal you must have your mother's functional allele. The father's allele was imprinted during spermatogenesis and is normally turned off.
A microdeletion in the PWS/AS critical area of 15 will cause PWS if it is inherited from the father and will cause AS if inherited from the mother. Likewise, whether or not a mutation in the gene for PWS or AS will cause the disorder depends on the parent of origin.
UPD is not unique to PWS and AS. We know that it can and does arise in other situations. The mechanisms of UPD are:
1. One parent's gamete contributes both chromosomes of a pair (heterodisomy) MI or both chromatids of a chromosome (MII) and the other parent contributes none of that chromosome.
2. Trisomy rescue. A zygote starts out as a trisomy and loses one chromosome of the three.
3. Monosomy rescue. A zygote starts out as a monosomy and during mitosis a non disjunction occurs giving rise to a diploid cell line.
4. May be the consequence of a de novo Robertsonian translocation where the "other" parents normal chromosome is lost (a trisomy rescue of the zygote). UPD may or may not cause a problem. If there are genes that are usually imprinted on the chromosome involved, one can expect problems.
Isodisomy is more apt to cause problems when it occurs than heterodisomy because it results in homozygosity for all genes on that chromosome. There was a case of cystic fibrosis (CF) in a child whose mother was a carrier but not the father. It was then shown that the child who had a 46,XX karyotype had inherited both copies of her chromosome 7 from her mother. She also showed short stature which could be due to imprinted genes. Subsequently, other similar cases have been documented leading to the speculation that UPD may not be unusual. Because imprinting of many genes during meiosis no doubt occurs, many hard to diagnose disorders may be due to UPD. Partial UPD has also been found when structural rearrangements are present.
Prader Willi and Angelman are examples of imprinting.
There are situations where the placenta is trisomic and the fetus is diploid but it had heterodisomy. As mentioned earlier, UPD, can arise from either trisomy or monosomy rescue. In a case of trisomy 16 rescue, the mother was heterozygous for two "markers" (RFLPs) on chromosome 16 and the father was homozygous for another marker. The placenta which was trisomic and showed all three markers. The fetus showed the same two markers found in the mother and none from the father. Therefore, the "normal" diploid fetus was actually heterodisomic for chromosome 16. Since there were both of the mother's markers, the non disjunction occurred in maternal meiosis I. (If only one of the mother's markers had shown up but in a double dose, and none of the father's, then it would have been isodisomy and due to a maternal meiosis II error.)
Trinucleotide Repeats (TNRs)
Fragile X syndrome (a.k.a. Martin Bell syndrome) is the most common cause of inherited mental retardation. The frequency in males is 1/1200 and 1/2500 in females. In addition to mental retardation, the clinical diagnosis includes an elongated face, large ears, large testes and, sometimes, autism. The earliest laboratory confirmations involved growing cells in folate deficient media which allowed one to see a fragile site cytogenetically on the X chromosome (Xq27.3) in 2-25% of metaphases in affected males. FRAXA is the name of the gene locus and FMR1 is the name of the gene involved. Fragile X was the first of what is now a number of disorders known to be associated with trinucleotide tandem repeats. Normal people also have the trinucleotide repeats but they have fewer repeats. When the number is expanded beyond a certain number characteristic for that gene, it causes a problem in the gene expression.
In 1991, this new kind of mutation was discovered, trinucleotide repeats (TNR), tandemly repeated 3 bp units. Normal people have these repeats but in small numbers, 5-50 depending on the disorder. The disorder results from the unstable expansion of the TNR (40-2000) in affected individuals, usually only in one allele. There are 2 classes of these disorders, the small scale expansions and the large scale expansions. There are 14 disorders which have been described thus far including: Fragile X (XL), Huntington (AD), Spinal Cerebellar Ataxias (SCA-there are several on different chromosomes; they are an example of locus heterogeneity); Myotonic dystrophy (AD), Kennedy disease (XL-androgen receptor gene) and Friedreich ataxia (AR-TNR in an intron). The genes involved are usually expressed in a variety of tissues. Researchers are looking at the functions of the normal gene products and the mechanism of expansion and instability of the repeats.
Except for Friedreich ataxia, these disorders show a phenomenon called "anticipation." This term referred to the fact that the disorder worsens with each succeeding generation. This is due to "allelic expansion" in meiosis (and sometimes mitosis) which can occur in either the male or female parents depending on the disorder. Contraction can also occur. Neither mechanism is well understood. The interjection of a slightly different nucleotide triplet within the repeat seems to stabilize the repeat (e.g., a CAA within the CAG repeats). Parent of origin effects are seen in most of these disorders in the sense that the expansion is more likely to occur in meiosis in the mother, Fragile X and Myotonic Dystrophy, and in others, Huntington and Kennedy, the expansion is more likely in meiosis in the father.
Many of the small scale expansion disorders, such as Huntington and Kennedy, exhibit a dominant gain of (mal)function (a negative effect). CAG codes for gln (Q) an uncharged polar amino acid. These repeats are expressed if within an exon and the increased number of gln (Q) somehow changes the functioning of the gene product. These smaller expansions with CAG repeats are neurodegenerative disorders, they have poly Q tracts in the protein, and they have a later onset than the larger scale repeat disorders. The greater the number of repeats, the earlier the age of onset. There is little or no somatic mosaicism of these expansions. The expansion is probably meiotic and can be seen in the sperm of the (unaffected) father. Mice, fortunately have many genes evolutionarily related to ours so we can construct KO (knock out) or null mice which do not have the functional gene. These KO or null mice do not show the same symptoms as the humans with the pathological number of TNR, therefore, the disorder is not due solely to a loss of the normal gene product. The disorder is due to the gain of a malfunction.
Huntington disease is an AD poly Q disease with CAG repeats within the first exon. The product of the gene is a large protein of unknown function called huntingtin. It is expressed in a variety of tissues in the cytoplasm. Since KO mice die because of an excessive amount of apoptotic cell death, huntingtin may be an inhibitor of apoptosis. The expansions to even larger numbers of TNRs occur in spermatogenesis. Normal is 9-30 repeats; with more than 42 repeats one can see cellular protein aggregates; and more than 62 repeats results in juvenile onset.
Huntington Disease was described in 1882 and called Huntington Chorea. The pattern of inheritance was described as autosomal dominant. The symptoms are due to the loss of neurons in the caudate nucleus (the acetylcholine neurons are not affected). Huntington Disease was found to be common in a community in Venezuela and was studied there in depth. A haplotyping approach using micro satellite markers was used and one third of the population with HD had a common haplotype from a common ancestor. The prevalence of the disorder in this Venezuelan community is due to what is known as a "founder effect." A European sailor is believed to have jumped ship and to have taken up residence there. He possessed the gene and passed it on. Because the community was small and inbred, the gene became common. It is a true dominant (AD) because homozygotes and heterozygotes manifest the disease to the same degree. The homozygous condition is not more severe nor does it result in an earlier onset. The gene was first located on 4p by positional cloning and later the CAG repeat was found. It was proposed that the problem was due to the lack of huntingtin but the loss of the gene in knock out mice proved lethal...so it must be a very important protein. Transgenic mice with "knock ins" of exon 1 with up to 150 glutamines show progressive nuclear mislocalization of huntingtin....it is not in the cytoplasm where it belongs. They also develop severe diabetes, brain shrinkage and die but there is no neuronal loss as in humans. Antibodies specific to huntingtin bound to inclusions in patients cells. The intra nuclear inclusions occur in neurons and other tissues as well. The poly glutamine proteins show altered mobility and a capacity to aggregate. Several proteins, many of which are involved in protein trafficking or RNA processing, are known to interact with the N terminal portion of huntingtin. There are several levels of intervention being pursued. One possibility is to find a compound that prevents the poly glutamine tract from acting to form inclusions.
Kennedy's disease (SBMA) is XL and only males are affected. It is caused by an expansion of a CAG TNR within an exon of the androgen receptor gene. The normal range of repeats is 12-34, affected have 40-62. 18 or less repeats have been associated with prostate cancer. African American men have fewer repeats and more prostate cancer. The repeats modulate the sensitivity to androgens.
Large scale expansion disorders such as DM (myotonic dystrophy), FraX, and FA (Friedreich ataxia) show early onset and also somatic mosaicism. In these disorders there is a loss of function with no protein being produced. The TNRs are in non coding regions. The repeat in FraX is in 5' UTR, in the 3' UTR in DM and in an intron in FA. There can be as much as a 20X increase in TNR size in one generation. The expansion occurs in a premutation (40-200) mother in FraX, and while there is more maternal expansion in DM there is also expansion in the male parent meiosis. Reduction of the number of TNRs is rare for DM and FraX.
Baby and young girl with Myotonic Dystrophy
Myotonic Dystrophy Diagnosis by Southern
Myotonic Dystrophy Diagnosis by PCR
FraX males have 200-2000 repeats at the 5' end of the gene in the promoter region. In Fragile X the larger number of CGG repeat can cause hyper methylation of the CpG island in the 5' UTR region of the promoter resulting in shutting off of the gene. This disorder is due to lack of the gene product. A normal male with a premutation is called a normal transmitting male (NTM). He will not have affected children since expansion does not occur in spermatogenesis (due to inactivation of his X in meiosis?). An NTM can pass his premutation to his daughters in whom it can expand. If the expansion is on the small side of abnormal, there is a normal amount of mRNA but the 5' end of the mRNA gets "stuck on the 40 S subunit of the ribosome and "stalls." No protein is made. It is believed that the FMR protein is an RNA shuttling protein that takes RNA through the nuclear pore to the ribosomes (it has a nuclear localization signal sequence). KO mice have abnormal dendritic formation.
Mytonic dystrophy (or dystrophia myotonica, DM) is the most common adult muscular dystrophy in humans. It has a complex, dominantly inherited pathology caused by a the triplet repeat, CTG, expansion in the 3' untranslated (UTR) of the serine-theonine kinase encoding DMPK gene. In myotonic dystrophy (DM) there is a CTG repeat in the 3' UTR of the DMPK gene (myotonin protein kinase) which is expressed in many tissues. The "expanded" mRNA stays in the nucleus and forms nuclear aggregates. Therefore, there is an insufficient amount of the gene product. The expansion occurs more in oögenesis. The myotonin phosphokinase (PK) is a signal transduction pathway protein and when it is not produced in sufficient quantities the pathway is down regulated magnifying the effect of the mutation and resulting in an AD phenotype with early onset of the disorder. Certain aspects of DM have been accounted for in previous mouse models of DM, apart from the Myotonic (hyper excitability) of the skeletal muscle. Recent research (2001) indicates that this characteristic of DM is not mediated by the DMPK gene itself but a by a toxic gain-of-function effect caused by the expanded CUG repeat in the mutant DMPK mRNA. New research into DM using mice suggests that the toxicity of the DM repeat is due to the CUG repeat in the mRNA which causes it to be retained in the nucleus where it is thought that it might interfere with RNA processing or interact with double-stranded RNA-binding proteins.
Friedreich ataxia is an AR loss of function disorder with GAA repeats in an intron. The protein product of the gene is called frataxin, its function is unknown at this time but it is expressed in a variety of tissues. The expansion causes a reduction in mRNA and, therefore, frataxin. in FA there is evidence of "contraction" of size from parent to child, especially in the male parent. The mutation appears to cause a mitochondrial defect. The normal gene product is localized to mitochondria (it has a mitochondrial signal sequence which is later cleaved off). Since the TNR is associated with a particular haplotype, there is a Founder effect (also true for DM). Recently, there is evidence of TNR in bipolar disorder and schizophrenia. CAG repeats at the 5' end of a gene coding for a tetrameric Ca++ activated K+ channel in neurons. The protein has 6 trans membrane domains = Ca++ pore, with the N and C terminus intracellular. The N terminal poly Q may interfere with the association of the tetramers. It maps to the 22q11-q13 critical region for bipolar disease and schizophrenia.
| DISORDER GENE NAME LOCUS PROTEIN |
TNR (amino acid) |
LOCATION IN GENE |
MODE OF INHERITANCE |
SIZE IN: *NORMAL *CARRIER *AFFECTED |
CHANGE IN mRNA OR PROTEIN | ANTICIPATION PARENT OF ORIGIN EFFECT |
MODEL |
| Fragile X FRAXA Xq27.3 FMR1 |
CGG | 5' UTR | XR | 5 - 52 43 - 200 230 - >2000 |
loss of function; methylation of CGG expansion shuts off transcription | maternal also mitotic instability |
KO mouse: macroorchidism, learning disabilities, hyperactive |
| SCA1 6p23 ataxin-1 |
CAG (gln) |
Exon 8 | AD | 6 - 39 -------- 41 - 81 |
abnormal ataxin aggregates in brain cells; removal of defective protein is impaired; interrupted repeat less likely to cause problems | paternal | Transgenic mice show Purkinje cell death with neurodegeneration and ataxia. Mouse gene has no CAG repeat role in cerebellar and vertebral column development. |
| SCA2 12q24 ataxin-2 |
CAG (gln) |
Exon | AD | 15 - 29 -------- 35 - 59 |
cytoplasmic protein | paternal | Mouse homolog has no poly Q tract. Gene expressed in most tissues including Purkinje cells and other CNS neurons |
| SCA3 Machado Joseph Disease SCA3 14q24.3-q31 ataxin-3 |
CAG (gln) |
Exon | AD | 13 - 41 -------- 62 - 82 |
nuclear protein; misfolding; nuclear inclusions; dosage effect | paternal | Transgenic mice with repeat are ataxic. Drosophilia with polyQ have nuclear inclusions and late onset cell degeneration especially in neurons. |
| SCA6 19p13 1A-voltage dependent Ca+2 channel subunit | CAG (gln) |
Exon | AD |
6 - 17 ------- 21 - 30 |
gain of (mal)function | none | |
|
SCA7 3p21.1 ataxin-7 |
CAG (gln) |
Exon | AD |
4 - 35 ------- 37 - 200 |
nuclear localization signal; associated with nuclear matrix and nucleolus | paternal | |
| SCA8 13q21 | CTG | 3' UTR |
AD
(confusing pedigrees due to lack of correlation with repeat size) |
16 - 50 normal 51 - 109 unknown may have increased risk 110 - 249 likely to cause ataxia 250+ unknown may not cause ataxia |
gene product is an RNA molecule not a protein; it partially overlaps with a gene in the opposite orientation thus it codes an "anti sense" RNA which can interfere with the function of the "sense" gene. |
> maternal < paternal |
|
|
Myotonic Dystrophy DM 19q13.2-q13.3 myotonin protein kinase |
CTG | 3' UTR | AD |
5 - 37 50 - 80 mildly affected >2000 |
RNA dominant mutation model: loss of function of 3 genes; may interfere with processing and export of other RNAs from nucleus |
>> maternal > paternal also mitotic instability |
KO and transgenic mice develop late onset progressive skeletal myopathy. |
|
Kennedy Disease SBMA AR Xq11-q12 androgen receptor |
CAG
(gln) |
Exon 1 | XR (expression in males only) |
12 - 34 -------- 40 - 62 |
repeats not in hormone or DNA binding domains; short repeats assoc. with risk for prostate CA; repeat length relative to age of onset of breast CA | paternal | transgenic mice with 45 or 66 repeats are normal; with 122 they are abnormal |
|
Huntington Disease HD 4p16.3 huntingtin |
CAG
(gln) |
Exon 1 | AD |
6 - 37 ------- 35 - 121 |
neuronal pathology due to poly Q fragmented protein aggregates; associated with HIP-1 and other proteins; cytoplasmic and in many tissues. Collects in nucleus. | paternal | KO mice embryonic death; transgenic mice with only exon 1 and repeat and whole gene are good clinical models |
|
Friedreich Ataxia FRDA X25 9q13-q21.1 frataxin |
GAA | Intron 1 | AR; 95% are homozygous for repeat mutation |
7 - 21 -------- 200 - 1700 |
no mRNA; loss of function of frataxin, a mitochondrial protein; accumulation of Fe | contractions > expansions; no anticipation | severe defect of mitochondrial respiration in KO yeast; loss of mtDNA, and elevated intra mitochondrial Fe; no mouse model |
|
DRPLA (Haw River Syndrome) 12p13.31 atrophin-1 |
CAG (gln) |
Exon 5 | AD | 7 - 23 ------- 49 - 88 |
dosage effect; intra nuclear protein |
> paternal
< maternal |
transgenic mice good models for investigating the molecular mechanisms of instabilities of CAG repeats. |
To summarize:
If the TNR is in an exon, extra amino acids are added to the protein product: (CAG)n = (gln)n or (poly Q). The result is a dominant negative effect because of a malfunctioning gene product. If the TNR is in a non coding region, gene transcription or regulation is affected. Friedreich Ataxia is the only autosomal recessive (AR) found thus far, the others are autosomal dominant (AD) except for Kennedy Disease which is X linked (XL).
The repeat length can change from generation to generation. The phenomenon known as "anticipation" is due to an increase in the length. In general, the longer the repeat, the earlier the onset and the more severe the disease. The same repeat can lead to variable clinical expression (e.g., DRPLA and Haw River Syndrome) which may be due to repeat size differences.
The expansion or contraction of a TNR occurs during meiosis and changes in length also occur during mitosis in some of the disorders. There is usually a "parent of origin" effect with the expansion occurring more frequently in the male or female parent depending on the disorder. Non triplet repeat sequences within a TNR can stabilize its length. TNR instability maybe due to changes in the secondary structure of the DNA which create polymerase pause sites thus facilitating slippage or sister chromatid exchange.
The TNR genes are often expressed in a variety of tissues but the effect is more severe in the CNS. The effects are often due to the interference of an abnormal gene product...a gain of (mal) function. Some gene products are nuclear proteins, one (frataxin) is a mitochondrial protein, some form aggregates.
Knock out (KO) and transgenic mice serve as models to study these disorders. Yeast and drosophila models also exist.
Diagnosis of a patient thought to have symptoms associated with a TNR disorder can be done using DNA analysis. Presymptomatic or predictive testing as well as prenatal diagnosis (CVS, amniocentesis or preimplantation genetic analysis) is also possible if there is a positive family history or confirmation of an affected relative. However, it is not recommended that anyone under the age of 18 years be tested unless they are showing symptoms. It is also required that there be a psychological evaluation and at least two genetic counseling sessions prior to collecting blood for any presymptomatic testing. An informed consent must be obtained for anyone undergoing testing of any kind.
Direct mutation analysis to determine the allele sizes is carried out by using PCR across the repeat region if the repeats are in the smaller ranges (those in the exons). Both the normal sized and expanded TNR alleles will be detected by this type of analysis. However, Southern analysis is used to identify the large sized expansions found in Fragile X and Myotonic Dystrophy which may not be detected by PCR. The methylation status of the TNR in Fragile X is done by using Eco R1 and BssH II restriction enzymes. The price is about $250 - 300 per sample or an SCA Panel for 1,2,3,6,and 7 costs $950. The accuracy is 95 - 99% detection.
Mitochondrial Inheritance
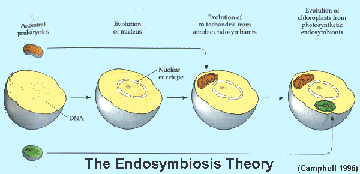
Mitochondria are believed to have originated from an endosymbiotic event in early eukaryotic cell origins. A mitochondrion closely resembles an aerobic bacterium and the eukaryotic cell now relies on this organelle to be the primary producer of ATP which drives all cellular functions. The mitochondria has an outer membrane which resembles the other cytoplasmic membrane of the eukaryotic (host) cell and an inner membrane which more closely resembles the prokaryotic (bacterial) inner membrane where the oxidative phosphorylation enzyme complexes are found. Mitochondria, like prokaryotic cells, have double stranded "naked" circular DNA (mt DNA) which codes for its own 22 tRNAs, 2 rRNAs, and 13 proteins that form part of the oxidative phosphorylation complexes. These RNAs are transcribed and the proteins translated in the mitochondrial matrix. However, the nuclear DNA (nDNA) of the cell also codes for many of the proteins in the enzyme complexes. These nuclear gene products are translated in the cell cytoplasm on free ribosomes and have signal sequences which send them to the mitochondria.
Mitochondrial disorders may be due to mutations in nDNA or in mt DNA. One can usually tell them apart by the pattern of inheritance. They both affect ATP production and, therefore, those tissues most dependent on ATP. Mutations in nDNA can interfere with the transport of mitochondrial proteins or iron into the mitochondria or between compartments, mtDNA replication, as well as the OXPHOS complex proteins themselves. Patterns of inheritance of mutations in nDNA that affect mitochondria include AD, AR or XL.
Some of the mitochondrial disorders are expressed only in conjunction with a nuclear gene mutation. The two locus model applies to LHON which is expressed only if the person inherits a mutant XL gene and sensorineural deafness which is expressed only in the presence of two doses of an AR expressed gene. Interestingly, both of these traits are associated with homoplasmy (all of the mitochondria in all cells are mutant or homogeneous) whereas most mitochondrial disorders show heteroplasmy (there are varying proportions of mutant and normal mitochondria in the cells of the individual).
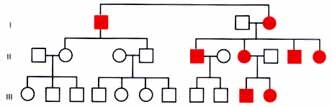
Mitochondrial inheritance is exclusively through the mother. This is because all of the mitochondria in the zygote come from the egg cytoplasm. The sperm contributes only its nucleus. (If an occasional paternal mitochondria were to be included it would be diluted out by the large numbers of mitochondria present in the egg.) Pedigrees will show both male and female affected but only the females pass the disorder on. The pedigree can look superficially like an XR pedigree but there is no male transmission through carrier daughters.
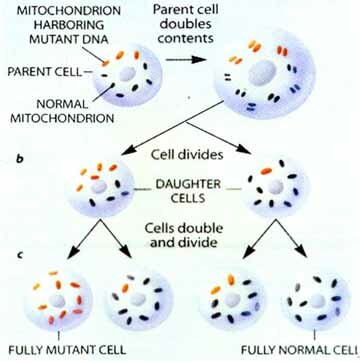
The human primary oocyte has about 100,000 mitochondria but loses most of them during maturation. The result is a stage in which there are between 10 and 100 mitochondria. During cleavage of the zygote, the number is built up to about 10,000 mitochondria per cell. The severe reduction of mitochondria in the oocyte is referred to as a "bottleneck". If a mutant mitochondrion is included in the 10 to 100 in the oocyte there is a chance that it will be over represented in the cells of the embryo. Mitochondria are randomly distributed into daughter cells, thus by chance alone some cells may receive more or less of the mutant mitochondria. Those tissues that receive a larger amount of defective mitochondria will have a lower ATP production. If the tissue is one which requires a large amount of energy such as cell of the nervous system, muscles, kidneys, etc., the person with the mutant mitochondria will be affected. The term heteroplasmy is used to describe the situation where a cell or tissue contains both mutant and wild-type mitochondria. The term homoplasmy describes the situation whereby all the mitochondria have the same genome, either wild-type or mutant.
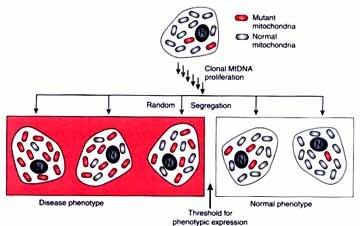
Sporadic cases of mitochondrial diseases are due to a new mitochondrial mutation in maternal mitochondria. The new mutation may be present in a minority of her cells but if they are in some of her primary oocytes she can pass them on. Therefore, an (apparently) unaffected mother can pass on mutant mitochondria in higher numbers than were present in her since the formation of an egg represents a "bottle neck" where the mutant mitochondria can be disproportionally included in the egg as compared with her other cells. Then these mutant mitochondria can multiply to produce a greater proportion of the 1000's of mitochondria in the sporadic cases known. A two locus model has been proposed to explain the inheritance of LHON--mitochondrial inheritance coupled with an X-linked gene explains the lack of affected offspring of some affected females, the slight excess of males, the later onset in females, and the pattern of affected females in pedigrees is accounted for by unfortunate X chromosome inactivation in females who are heterozygous for the X-linked gene.. Affected people are homoplasmic for the mt mutation most of which are due to one of 2 or 3 point mutations in respiratory chain proteins. There is more than one mt mutation that can cause LHON (allelic heterogeneity); however, in any one family all affected family members will have the same mutation. For the most part LHON is a tissue specific mitochondrial disorder of the eye although cardiac dysrhythmia is frequently associated with the optic neuropathy but there is no skeletal muscle involvement. Less variation in this mt disease; some variation in age of onset and severity.
Sensorineural deafness.
One form of sensorineural deafness is due to homoplasmic mt mutation coupled with an AR gene.
Pearson marrow-pancreas syndrome.
Pearson Marrow-pancreas syndrome is a severe anemia in infancy with exocrine pancreatic dysfunction. Mt DNA deletions have been demonstrated. Mutations in nuclear genes coding for oxidative-phosphorylation subunits also cause mitochondrial disorders. Leighs disease (AR) is an invariably fatal degenerative brain disease. It is caused by a deficiency in a Complex IV subunit (cytochrome oxidase). The patients have encephalopathy with seizures, ocular and respiratory problems. There is an interesting AR disorder called the Mitochondrial Depletion Disease. The defect is due to a defect in the mt DNA replication whose replication enzymes are all coded for by nuclear genes. One finds mitochondria with no mt DNA. In one family 2 sisters died of the mitochondrial myopathy and a cousin from liver dysfunction. The defect is in the regulation of the mt DNA copy number.
Environmental factors can mimic these disorders (phenocopies). Drugs that interfere with mt DNA replication such as AZT which is a base analog can cause myopathy, RRF and weakness. Some antibiotics are known to cause deafness by damaging mitochondrial function.
Some interesting questions are: Why do only dup/del mutation and not base substitution mutation in mt tRNA cause CPEO? Why is only the optic nerve and sometimes the heart affected in LHON if all cells are homoplasmic? Why do you find homoplasmy only if nuclear genes are involved?
Attempts at therapy (RX) try to boost ATP production by giving electron shuttling substances such as coenzyme Q (which also stabilizes the inner mt membrane), succinate (substrate), riboflavin (FAD, coenzyme), Vitamin C and Vitamin K. The last four all act as surrogate e- transport molecules in specific area of the electron transport system (ETS).
Veterinarians are aware of a mitochondrial disorder in Holstein cows. It is a heteroplasmic mt DNA point mutation with considerable variation between sibs.
New areas of interest that appear to be associated with mitochondrial defects are Alzheimer, Parkinson and aging.
Mt DNA has also played an important part in exploring human evolution. The "mitochondrial Eve" is believed to have originated in Africa. Mt DNA has the advantage of being haploid and only inherited from one parent (the Y chromosome has also proven useful for the same reason). The timing of evolutionary divergence is done by following the number of mutations that have accumulated, a DNA clock. The technique has proven useful in looking at the evolutionary divergence of other closely related species such as dogs and wolves.
Comparison of the sequence of mt DNA from the skeleton believed to Czar Nicholas, the exhumed bones of his younger brother, and the mt DNA from two living relatives showed a rare mt DNA mutation present heteroplasmically in the two brothers' skeletons and homoplasmically in the two living relatives. There are many more examples of human evolution studies and forensic uses of mt DNA.
Mitochondrial DNA Diseases
This table lists only some of the disorders that can be caused by mutations in mitochondrial DNA. Certain of these conditions can also be caused by nuclear mutations or other processes that hinder mitochondrial function.
Disorders and Disorder Features
| Alzheimer's disease (in some cases) | Progressive loss of cognitive capacity |
| CPEO (chronic progressive external ophthalmoplegia) | Paralysis of eye muscles and mitochondrial myopathy |
| Diabetes mellitus (in some cases) | High blood glucose levels, leading to various complications |
| Dystonia (some cases) | Abnormal movements involving muscular rigidity; frequently accompanied bv degeneration of the basal ganglia of the brain |
| KSS (Kearns-Sayre syndrome) | CEO combined with such disorders as retinal deterioration, heart disease, hearing loss, diabetes and kidney failure |
| Leigh's syndrome | Progressive loss of motor and verbal skills and degeneration of the basal ganglia; a potentially lethal childhood disease |
| LHON (Leber's hereditary optic neuropathy) | Permanent or temporary blindness stemming from damage to the optic nerve |
| MELAS (mitochondrial encephalomyopathy, lactic acidosis and strokelike episodes) | Dysfunction of brain tissue (often causing seizures, transient regional paralysis and dementia) combined with mitochondrial myopathy and a toxic buildup of acid lactic acidosis and in the blood |
| MERRF (myoclonic epilepsy and ragged red fibers) | Seizures combined with mitochondrial myopathy, may involve hearing loss and dementia |
| Mitochondrial Myopathy | Deterioration of muscle, manifested by weakness and intolerance for exercise; muscle often displays ragged red fibers, which are filled with abnormal mitochondria that turn red when exposed to a particular stain |
| NARP (neurogenic muscle weakness and retinitis pigmentosa) | Loss of muscle strength and coordination, accompanied by regional brain degeneration, ataxia and deterioration of the retina |
| Pearson's syndrome | Childhood bone marrow dysfunction (leading to loss of blood cells) and pancreatic failure; those who survive often progress to KSS |
SCIENTIFIC AMERICAN August 1997
