BIO 442 MENU
syllabus 
1 - genome
2 - mutate
3 -cell cycle
4 - karyotype
5 - chromoabn
6 -sex-determ
7 -prenatal
8 - mendelian
9 - complex
10 - non-trad
11 - clinical
12 - newborn
13 - teratog
14 - linkage
15 - DNA prof
16 - quanti
17 - links
18 - quizzes
(full title of lecture appears in status bar on the top or at the bottom of your window)

Biology 442 - Human Genetics
Sexual Differentiation
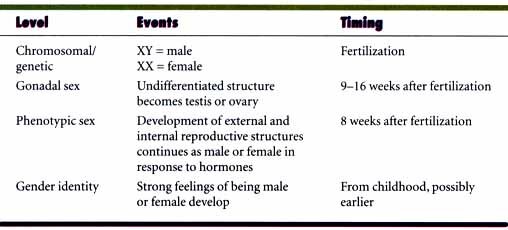
Sex means different things in different contexts. Sex in biology usually refers to the recombination of genes to produce genetically variable offspring. Sexual reproduction is different from asexual reproduction which results in identical cells or individuals except for mutations. Asexual reproduction is commonly found in prokaryotes although they, too, have several mechanisms of recombination of genes (conjugation, transformation, transfection, etc.). Fungi, protozoa, plants and some animals reproduce asexually by mitosis as well as sexually via meiosis. Even self fertilization or parthenogenesis is preceded by meiosis whereby the parental genetic material is "shuffled" during meiosis by random segregation of chromosomes and crossing over between homologs. Some lizards reproduce by parthenogenesis and there are no males. Queen bees and worker bees are diploid and develop from the queen's fertilized eggs but male bees (drones) develop from unfertilized eggs and are therefore haploid and do not survive long. They produce sperm (by mitosis) to fertilize the queen bee's eggs. These are variations on the "usual" but are still sexual mechanisms.
Sex: Chromosomal, Gonadal, Internal and External Plumbing, Psychosocial
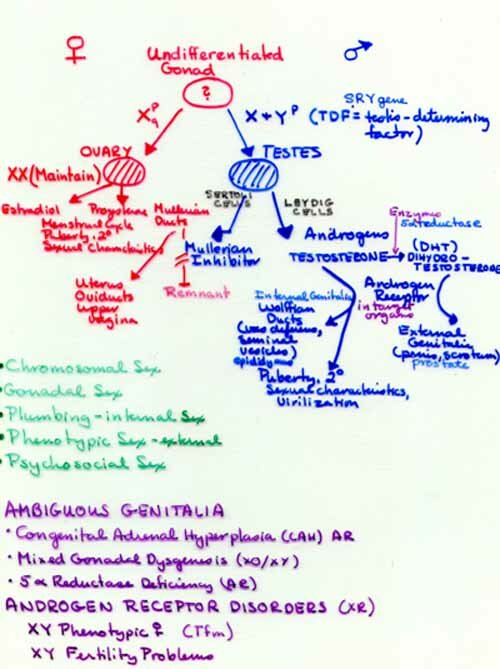
Before this discussion of sexual development in humans (mammals), we must first define what we mean by sex. There is chromosomal sex. Whether you are XX (female) or XY (male) with regard to the sex chromosomes. There is gonadal sex. Do you have ovaries (female) or testes (male)? Do you have male or female internal plumbing? This concerns the ducts which guide the gametes or embryo out of the body. Females have plumbing derived from the embryonic Müllerian ducts: oviducts, uterus, and vagina. Males have plumbing derived from the Wolffian ducts: epididymis, vas deferens, urethra. Do you have male or female external plumbing or paraphernalia? This is sometimes referred to as "phenotypic sex." Externally, males have a scrotum and penis, females have labia majora and minora and clitoris.
A complex series of steps must occur in gonadal differentiation. A number of genes are critical to appropriate male genital development. SRY (sex determining region of the Y chromosome), a gene on the short arm of the Y chromosome, is a testis determining factor. The SOX9 gene is also important in male sexual differentiation. DAX1, an orphan member of a nuclear hormone receptor family located on the X chromosome, interacts with steroidogenic factor 1 (SF-1). Other genes involved in male gonadal differentiation include the tumor-suppressor gene WT1 (Wilms' tumor 1), and the Müllerian inhibiting substance gene (MIS) and its receptor, MIS-R.
There is also psychosocial sex or gender identification. This refers to whether we think of ourselves as male or female or do we behave and feel more like the "other" sex than we have been assigned phenotypically. This includes all forms of homosexuality. We will not go into this in detail in this course but there are a couple of things to keep in mind. Homosexuals are not homosexuals because of anything unusual about their chromosomes, gonads or plumbing. There is evidence that there are genetic factors, particularly in male homosexuality. Female homosexuality is somewhat more complex. There is also considerable evidence that the hormonal environment of the developing embryonic and fetal brain is important in sexual behavior. When testosterone is absent during development the sexual orientation is female. One of nature's experiments (congenital adrenal hyperplasia, CAH) causes an excess of androgens and when a female fetus is exposed to excessive amounts of androgens the result is that their behavior is often more "boy-like." CAH shows allelic heterogeneity and locus heterogeneity. The default sex in mammals is female. Only with clear orders from the SRY gene will an embryo become a male. Until week 7 both sexes appear similar even though their sex is determined at fertilization. The early period is the "indifferent" stage. Early in week 4 primordial germ cells from the yolk sac migrate to the gonadal ridges. During week 6 the germ cells become incorporated into the gonad. Two pairs of ducts form in both sexes; these are the mesonephric (kidney, Wolffian) ducts and the paramesonephic (Müllerian) ducts. The latter will form the Y shaped utero vaginal primordium in the female. In the male, the development of the Müllerian ducts is inhibited by anti-müllerian hormone (AMH) A.K.A. müllerian inhibiting substance (MIS) which is produced by the Sertoli cells of the testes. The production of AMH is controlled by at least two autosomal gene loci. One codes for the hormone and one for its receptor. Mutations in either gene can result in the persistence of Müllerian ducts in the male.
Wolffian Ducts
The Wolffian ducts develop under the influence of testosterone which is made by the Leydig cells of the testes. They are the progenitors of the internal male plumbing: the epididymis, vas deferens, seminal vesicles and prostate gland. Until week 9 the external genitalia of the two sexes are similar in appearance. Soon the genital tubercle elongates to form the phallus which develops into the penis and the urogenital folds fuse.
The Y makes the Guy
There's more to the Y than we thought!
TDF/SRY Gene
To develop testes, the embryo needs a Y chromosome with an intact TDF/SRY (testis determining factor/sex determining region) gene (Yp11.3) and it also needs an X chromosome. It is the TDF/SRY gene which is responsible for the initiation of the development of the undifferentiated gonad into the testes. The gene contains one exon and codes for a 204 amino acid protein with a 79 amino acid HMG box. The gene product is a putative transcription factor with a highly conserved 76 amino acid DNA binding domain, homologous to the so-called high mobility group (HMG) protein known to cause DNA bending and activate gene transcription. The remainder of the protein sequence shows divergence, this area is probably responsible for the selectivity of the 5' binding sites. Appropriately, the gene is expressed only in Sertoli cells of the primordial gonad shortly before testis differentiation. The TDF gene is believed to initiate the development of the testes by repressing an X-linked gene, the "Z" gene, which would otherwise direct the fetus toward female development. Homozygosity for mutations in the Z gene are believed to account for some XX phenotypic males. The AMH gene mentioned earlier is among the genes that get turned on by TDF/SRY. TDF/SRY belongs to a family of structurally related genes (SRY related genes = SOX genes) which have recently been identified.
Sex Reversal Disorders
Other rare sex reversal disorders will help elucidate the testes determining pathway. One such gene is the SOX 9 gene (17q24-25) mutated in campomelic dysplasia (CD) an AD lethal skeletal dysplasia in which an XY fetus may have female genitalia. The SOX 9 is the only SOX gene with 2 introns. Its protein has 509 amino acids with an HMG box. Also, some XY females have a duplication of the DSS (dosage sensitive sex reversal) locus (Xp21.2-p22). Two active copies of this gene overrides the TDF/SRY signal.
The X and Y Chromosome

To become a fully functional (reproductive) female you must have two X chromosomes. To become a phenotypic female you need only one X but you need both the p and q arms (Turner female) to be a fertile female. Some Turner females who are mosaics (46,XX/45,X) have ovaries with eggs. (Mice with only one X are fertile.) Ovaries develop unless the TDF/SRY gene is present. Estridiol produced by the fetal ovary is responsible for the maturation of the Müllerian ducts. Without testosterone, the Wolffian ducts which give rise to the male internal plumbing, regress in the female.
The Y chromosome is small, with poor gene content. About half of the q arm is heterochromatic and there are no genes there. Thirty percent of the chromosome is euchromatic. Except for the Yp arm, it does not recombine and is passaged only through males. Therefore, it is useful in human evolution population studies and DNA identity testing. Polymorphisms on the Y chromosome were used to identify Thomas Jefferson as the father of his slave Sally Henderson's children by comparing the Y chromosome of male descendents of the male offspring of his brother and the Y chromosomes of Sally's male children's male descendents. Thomas Jefferson had no male children by his legal wife so it was necessary to use his brother's descendents to determine relatedness of Sally Henderson's descendents to him. This strategy was based on the fact that Thomas Jefferson and his brother shared the same Y chromosome they inherited from their father.
The Y chromosome is believed to have evolved relatively recently from the X chromosome. There are several regions of homology between the X and Y. Although sex is determined by a number of different mechanisms in other organisms (e.g. environmental cues), several groups of organisms have evolved a "Y" chromosome. This is an example of convergent evolution. The male determining sex chromosomes all accumulate similar properties so that it appears there is a universal pathway. The mammalian Y which probably evolved from the X has also acquired genes from other chromosomes especially those that enhance male fertility. Currently, there are twenty known single genes. More are known but many are multiple, tandemly repeated copies and many are pseudogene copies of active genes on the X chromosome (e.g., Xg and STS). The Y also has many inversions. Some of the genes on the p arm are: SRY (sex determining region); ZFY (zinc finger protein); RPS4Y (ribosomal protein S4); TSPY (testes specific protein); AMELY (amelogenin a highly conserved protein in dental enamel which has a distinguishable X homolog, they are used in sex identification of dead bodies); and on the q arm: DAZ (deleted in azoospermia probably originated from chromosome 3, homologs in the mouse and drosophila are on an autosome); AZF1 (azoopermia factor 1); UTY (ubiquitously transcribed tetratricopeptide repeat); RBM (ribosomal binding motif proteins).
The functional genes on the Y chromosome fall into two classes: Class 1 are X homologs and Class 2 are Y specific. The Class I genes have an X homology, are single copy and are ubiquitously expressed. They are housekeeping genes and may have testes specific transcripts, too. Class 2 have multiple copies, except for SRY, and are expressed in the testis. They function in spermatogenesis and these genes are in the repetitive regions of the Y chromosome. Class 1 genes survived decay because of their housekeeping role. Class 2 were acquisitions of male beneficial functions. They are Y specific, amplified on the Y and play an essential role in male fertility. De novo Yq deletions account for 10% of all male infertility cases involving spermatogenesis failure.
The X and Y are thought to have diverged in "blocks" due to inversions on the Y. There are 5 regions of the Y chromosome: Yp has the PAR in which there is no sequence divergence between the X and Y. Then comes region 1 with 10% sequence divergence, 2 with 30%, and 3 with 60%. In pro-simians, regions 1,2, and 3 are still PAR. Yq has region 4 with 90% and 5 with 100% sequence divergence and the heterochromatin region. Region 5 diverged 300 MYA when dinosaurs were around (Mesozoic-Triassic). If regions 3 and 4 of the Y are present in a Turner female, (XYp-) she can develop gonadal blastoma.
Over time the Y genes have "decayed" by reduction of expression and disruption of the ORF. The X genes without a Y homolog are the ones that get inactivated. The Xq is the ancestral X and it is where the most LINES are and where the genes are already inactivated. Those X linked genes with functional or "dying" Y homologs do not get inactivated (since it takes awhile for evolution to catch up and inactivate the X homolog). The Xp is the "younger" part of the X and as the Y linked genes are lost, the X "has to" inactivate the X homologous gene. X inactivation evolved on a gene by gene basis and the inactivation of X genes is preceded by "decay" of its Y homolog. Haploinsufficiency of XY genes may contribute to Turner Syndrome. (The Class I genes are sometimes referred to as "Turner genes.") The Class II Y specific genes for which there is no X homolog, may have come from autosomes. They all deal with male fertility. CDY, a Class II gene on the Y, codes for a testes specific histone acetyl transferase (HAT). It is a retroposed copy of its autosomal homolog, CDYL (L = like) and was acquired about 30 - 50 MYA. The mouse has no Cdy (mouse genes are specified by lower case letters) on its Y and the Cdyl is expressed in all tissues. In primates, the autosomal CDYL takes care of housekeeping function in all tissues except the testes which has the Y CDY gene expressed. HAT activity is necessary for the displacement of histones which are replaced by protamines in mature sperm chromosomes. Sperm use protamines in place of histones since protamines package DNA more compactly.
The X and Y chromosomes pair during spermatogenesis. This pairing is in the pseudo-autosomal (PAR) regions of both chromosomes. (The genes in the PAR are not inactivated.) Slippage sometimes occurs during the pairing and since the TDF/SRY locus is close to the border of the PAR, the TDF/SRY gene may be translocated to the X. This results in an X with the TDF/SRY locus and a Y without a TDF/SRY locus. Therefore, you can have XX males and XY females. 80 -90% of XX males have translocations of the TDF/SRY to one of their X chromosomes. So the remaining 10 - 20 % must represent mutations in genes other than TDF/SRY involved in testes determination. Some XY have deletions of the locus but some have mutations of the TDF/SRY/SRY gene. The mutations usually occur in the HMG box DNA-binding region. However, since these account for only 15-20% of XY it is believed that there are other genes in the testis determining pathway that can mutate and cause sex reversal, too. Because 9p- and 10q- can result in XY with dysgenic ovaries, some of these genes may be at those sites.
A poster at the 1998 ASHG meeting described a 45,X male who had inherited a paternal X from his father as evidenced by a study of the polymorphic locus DXS52. The father's X and Y had undergone an unequal crossing over event and the X had received the SRY gene from the Y. The zygote had lost the mother's X chromosome. Since he had the SRY gene on his X chromosome, the child was a male. In addition, the father's SHOX gene, which the boy had inherited on the X, was mutated. The SHOX gene is a gene that contributes to height. It is on the pseudoautosomal region of the X and Y chromosomes. Turner females are shorter because they are missing one SHOX allele. Individuals who are XXY, XYY, XXX are taller than XX or XY because of the additional SHOX genes. Because of the mutation in their SHOX gene, both father and son had a disorder known as Leri-Weill dyschondrosteosis. This is a (pseudoautosomal) dominant skeletal abnormality which results in a wrist deformity, mesomelic dwarfism, short stature, normal head and face and normal IQ.
Testosterone and Dihydrotestosterone
Testosterone produced by the testes and is responsible for the development of the internal male ducts derived from the Wolffian ducts. It is also responsible for secondary sexual characteristics and further virilization at puberty. Testosterone is converted to dihydrotestosterone (DHT) by the enzyme, 5 alpha reductase found in certain target tissues. DHT is responsible for the development of the external genitalia in males in early development. There is an AR condition whereby the activity of 5 reductase is lost. This condition results in ambiguous genitalia and the affected boys may be mistakenly raised as girls. At puberty these boys become virilized and develop normal secondary sexual characteristics. There is a community in the Dominican Republic which is inbred and where there is a "Founder Effect" for this mutation. The defect is relatively common and is referred to as "Huevos a doce" (pardon my French). Another AR genetic defect which results in ambiguous genitalia is congenital adrenal hyperplasia (CAH) about which we will have more to say later. It is a defect in the steroid synthesis pathway of the adrenal gland and it results in an excess of androgens. In female fetuses, their external genitalia may be masculinized and they may be mistaken for males because the clitoris is enlarged and the labia may be partially fused. Because the frequency is higher in some regions, some states include a test for CAH in newborn screening. Such ambiguous genitalia can now be corrected surgically at birth or soon afterward.
Androgen Insensitivity
Another genetic defect is the testicular feminization or androgen insensitivity syndrome. It is inherited in an XR pattern. The disorder is caused by a defect in the androgen receptor. The androgens, testosterone and dihydrotestosterone are steroid hormones. All steroid hormones enter the cell directly and are picked up by a receptor which ferries them into the nucleus and to the chromosomes (DNA). In the nucleus the hormone and its receptor act as a transcription factor. Neither testosterone or DHT can function without this receptor. This same gene is involved in one of the trinucleotide repeat (TNR) disorders, Kennedy disease. Many different mutations have occurred in this gene (allelic heterogeneity) and they are responsible for a wide variety of phenotypes. When the receptor is not functional, the result is the full Androgen Insensitivity Syndrome (AIS). AIS individuals present as normal appearing females who are tall (due to the Y chromosome genes) and thin with primary amenorrhea. When karyotyped they are found to be 46,XY and upon physical examination will have a blind vagina and internal testes. Like Turner females with testicular material, the gonads must be removed in an XY female because they have a high probability of developing gonadal blastomas. At the opposite end of the spectrum, those with milder mutations are normal appearing males with reduced fertility.

XY Androgen Insensitivity
The gene (TDF/SRY) responsible for the development of the testes is on Yp. Its role is to begin the cascade of gene products responsible for "maleness" beginning with the development of the testes. The inclusion of SRY in transgenic XX female mice will cause them to differentiate as males (see Passarge, p. 325). SRY belongs to a family of SOX genes (SRY-like HMG-box).These share an 80 amino acid domain with high mobility group (HMG) proteins that bind DNA at a 6 to 7 base consensus sequence. Mutations in the SRY gene can occur in the HMG box region. They can result in a female phenotype with XY gonadal dysgenesis with streak gonads (as in Turner syndrome). 80 - 90% of XX males have the SRY gene translocated to the X. However, since a few do not have the SRY gene, this suggests that autosomal genes are involved in sex determination. The many known SOX genes are highly conserved and many act as transcription factors in different developmental pathways. (SOX was named after HOX or homeotic genes which control the function of many genes during development.)
Both SRY and SOX9 (17q24-25) are required for testis determination. Mutations in SOX9 in an XY fetus can result in phenotype ranging from normal male to normal female with dysgenic gonads. It also causes a lethal condition, campomelic dysplasia, which is characterized by skeletal abnormalities as well as sex reversal in most XY individuals. SOX9 regulates chondrogenesis in mice. There are other autosomal genes involved in sex determination on chromosomes 9 and 10. 9p- and 10q- can result in XY with dysgenic ovaries. An infant has been described as having campomelic dysplasia with the following features: hydrocephalus, congenital heart defect, dysmorphic features and complete sex reversal--46, XY karyotype with complete female phenotype. The baby did not have campomelia, however.
SRY most likely acts as an inhibitor in the male sexual differentiation pathway rather than an activator. The SRY target is a "missing link" gene which is probably a suppressor gene that inhibits SOX 9 in females but which itself is inhibited by SRY in males. Such a double inhibition could also account for XX males with no SRY. A candidate "missing link" gene is the X linked DAX1 gene which suppresses testes determination when present in duplicate (DAX1 = DDS, dosage sensitive sex reversal), i.e., two active copies of DAX1 (Xp21.2-p22) override the SRY signal. DAX1 is also involved in adrenal differentiation. Another candidate for the "missing link gene" is the SOX3 gene which lies on the older, conserved part of the X chromosome. SOX3 is also expressed in the CNS.
The Anti Müllerian Hormone (AMH) or Müllerian Inhibiting Substance (MIS)
The anti Müllerian hormone (AMH) is one of the downstream genes regulated by the SRY/SOX pathway. Inherited as an autosomal recessive condition, a male fetus with two mutant AMH genes (or with mutations in the receptor for the anti-müllerian hormone will have persistence of the (female) Müllerian structures. At the same time, the XY fetus is developing all the normal male internal and external organs, he will also have vestigial female ducts. At some time later in his life this person may have what is thought to be a hernia and the surgeon will find the rudimentary female ducts within the scrotal sacs.
XY Females and XX Males
XY females can result from: 1. mutations/deletion of the SRY gene. 2.a deletion of the SRY gene by translocation. 3. duplication of the DSS (DAX1) gene on the X chromosome (2 copies override the SRY signal). 4. mutations in the SOX 9 gene. 5. Tfm/AI (testicular feminization or androgen insensitivity) is due to mutations in the XL gene for the androgen receptor. XX males can result from the addition of the SRY gene by translocation from the Y.
Mutations in the androgen receptor gene can result in a wide range of phenotypes from phenotypic female to infertile male. This is an example of allelic heterogeneity. Note that the phenotypes associated with mutations in the same gene but at different sites may not appear to be related at all clinically/phenotypically. This is in contrast to locus heterogeneity which refers to mutations in different genes at different loci which cause the same or similar phenotypes (genocopies). Steroid hormones enter the cell and bind to a receptor which carries them to the nucleus. The hormone-receptor complex then acts as a transcription factors turning other genes on or off. This is different from protein or peptide hormones which "knock" at the door of the cell by binding to receptors in the cell membrane. These protein or peptide hormones never enter the cell but instead rely on the receptor to begin a cascade of intracellular events which produce a response.
The androgen receptor has two binding sites, one for the androgens, testosterone and dihydrotestosterone (DHT), and another for DNA. The receptor binds the hormones in the cytoplasm after they enter. Together the receptor and hormone enter the nucleus where the complex acts as a positive or negative transcription factor for a variety of genes controlled by that hormone. The protein structure of the androgen receptor (AR) reveals a binding domain composed of a DNA binding site and a separate hormone binding site. When the mutation affects either site the regulatory activity of the receptor will be affected. If there is no activity, the result is the testicular feminization (Tfm) or androgen insensitivity (AI) syndrome. The resulting female has testes, no Müllerian ducts, no Wolffian ducts, and female external genitalia. The testosterone can be converted into estriol and the person becomes feminized. Tfm females are tall (both SHOX genes on the X and Y are working), have breast development, blind vagina, no pubic or axillary hair. Like Turner females, they are often diagnosed only after puberty when they are seen for primary amenorrhea. Mutations in the receptor may only cause less stable binding of hormone and thus produce a range of phenotypes including under virilized male, infertile male, etc. Allelic heterogeneity accounts for the various phenotypes associated with degrees of androgen insensitivity and a range of phenotypes due to differences in the binding at these two sites. Remember that Kennedy disease, a trinucleotide repeat disorder, is also due to mutations in the androgen receptor. The androgen receptor gene is on the X chromosome.
External genitalia have an intrinsic tendency to feminize and masculinization requires androgens be present prior to 12 weeks in fetal development. A derivative of testosterone, DHT (dihydro testosterone), is the primary virilizing androgen in the fetus for the male external genitalia. DHT is formed from testosterone by an enzyme, 5 alpha reductase, found in the end organ tissues. Because of an absent or abnormal enzyme T cannot be converted to DHT. Since the tissues involved in the external male structures respond most efficiently to DHT and not T, the external genitalia are predominantly female when only T is present. The result is ambiguous genitalia in male babies who are otherwise in possession of internal male paraphernalia. At puberty the child is virilized by increased testosterone and acquires male secondary sexual characteristics with some growth of the phallus. None of the affected males in published pedigrees have offspring. This is most likely due to lack of a functional penis and possibly the effects of un descended testes. The syndrome is inherited as an AR and is best described in several Dominican kindreds. Because brain receptors probably respond to testosterone, children often choose to be male at the time of puberty. If recognized early, infants can now be treated specifically with DHT with a good response.
Congenital Adrenal Hyperplasia
Congenital adrenal hyperplasia is a group of disorders all inherited as AR, caused by a deficiency of various enzymes involved in steroid synthesis. Several can be associated with genital ambiguity. Ninety-five percent of the cases of CAH are due to the deficiency 21-hydroxylase, which involves the gene for the adrenal "microsomal" cytochrome P-450 specific for steroid 21-hydroxylation. 21-hydroxylase deficiency has been associated with at least three different clinical presentations due to allelic heterogeneity--the loss of salt (no enzyme activity), the simple virilizing (2% enzyme activity), and the late onset (10 - 20% activity). The incidence is 1/5000 births with a carrier frequency of 1/35 making it the most frequent AR genetic disorder. In Yupik Eskimos of western Alaska, the incidence is 1/11. The late onset type accounts for 6-12% of hirsutism in women. CAH can also be caused by a deficiency of 17-alpha-hydroxylase. This gene CYP17 is on chromosome 10q24.3. This is an example of locus heterogeneity: same phenotype, different genotypes.
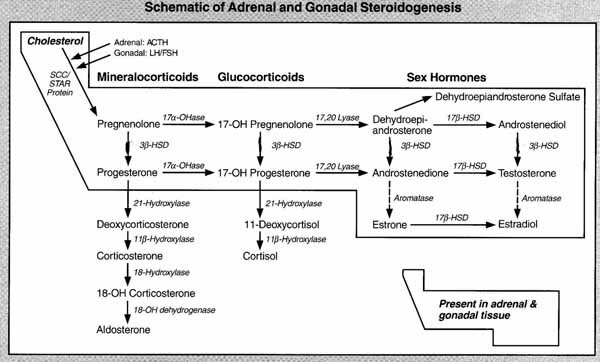
The over secretion of adrenal androgens in CAH, which begins at the third month of gestation, masculinizes the external genitalia of female fetuses, resulting in ambiguous (external) genitalia. The involvement is variable but may be severe enough to allow formation of a normal phallus. The gonads are normal ovaries and the internal plumbing arise normally from the Müllerian duct. Clues to the diagnosis include the absence of gonads in the scrotum, the presence of a uterus and elevated 17-ketosteroids. The gene for 21-hydroxylase (CYP21B) and its closely related pseudogene (CYP21A) are located on the short arm of chromosome 6 (6p21.3), separated by approximately 30 kb right in the middle of the histocompatibility loci. The two genes (CYP21 A and B) are very closely related in sequence, with about 97% to 98% homology. The arrangement suggests a tandem duplication of an ancestral unit early in evolution, probably due to an unequal cross over event. Unequal pairing between homologous chromosomes is facilitated by the very high level of sequence homology between the CYP21A and CYP21B. This can result in chromosomes that lack units or have additional units or fusion genes that are passed to sperm and egg cells in meiosis. Such misalignment and subsequent duplication and deletions are seen in other repetitive gene sequences such as the color vision genes on the X chromosome and the and globin gene complexes.
CAH can now be treated prenatally with dexamethasone from 9 weeks to term to obtain normalization of genitalia compared to prenatally untreated sibs. (See p. 331 in Passarge). Both the classical salt wasting type and the simple virilizing type have elevated 17-hydroxy progesterone. Further testing will show normal electrolytes in the simple virilizing type. Some states include CAH in their newborn screening panel (California does not). They measure 17-hydroxy progesterone in the blood. One difficulty with the newborn screen is that there may be an unacceptably large number of false positives from premature babies who may be normal but have high levels of the analyte.
CAH, like other enzyme deficiency disorders is AR because heterozygotes with one functional allele make 50% of the normal amount which is sufficient. This is an example of a gene dosage effect.
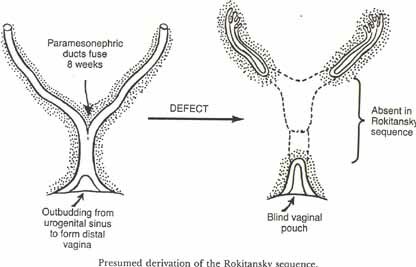
Müllerian Agenesis/Dysplasia
Müllerian agenesis or Müllerian dysplasia is a condition whereby a female has incomplete internal plumbing. Something goes wrong with the Müllerian duct formation. Normally one thinks of only males having anti müllerian hormone (AMH) but some is made in the early ovary. The function of AMH (a.k.a. MIS) is to repress Müllerian duct formation or persistence in the male. The hormone is a protein and, therefore, has a cell membrane receptor. Mutations in either the gene for the hormone or for the receptor can cause problems. If a male does not make a normal amount of the hormone or if he has a non functional or less functional receptor for the hormone, he will have remnants of the Müllerian duct structures (fallopian tubes and uterus). Females make some AMH is the ovary and if more is made or a more potent form is produced or if the receptor is mutated to be always "on" the female can have Müllerian agenesis. There is variation in the extent to which the Müllerian structures are absent. The Rokitansky sequence is the name often given to incomplete internal female plumbing. Most patients will have a blind vagina, some will have underdeveloped oviducts and a rudimentary uterus. One patient we recently saw had no internal plumbing and we are still looking for the ovaries which are probably there but very small (she has female secondary sexual characteristics). This patient is also a Duarte heterozygote for galactose-1-P uridyl transferase, the enzyme defective in galactosemia. The relationship is not clear but it has been found in several woman with Müllerian agenesis. She also was missing a kidney and had a flattened face which has also been associated with a Müllerian agenesis syndrome called a urogenital adysplasia. We had one patient with triple X and Rokitansky sequence.
AMH is coded for by a gene on 19q13 and the AMH receptor is on 12q13. Mutations in either gene cause the persistence of Müllerian duct syndrome (in males). The inheritance pattern for both is AR.

Psychosocial Sex or Gender Identification
Psychosocial sex or gender identification is a new area of scientific interest. At one time it was believed that gender identification depended only on social and environmental influences such as rearing, learning and individual choice. A book was written recently about a male twin whose circumcision ended in the burning mutilation of his penis. He was raised as "Joan" instead of John along with his twin, Kevin. Despite surgery. psychotherapy, and hormone treatment, Joan never felt like a girl. When the parents finally told Joan, a teenager then, the truth about how she became a girl, Joan is reported to have felt a sense of relief and soon after made the decision to have a sex change back to being a male. S/he had her breasts removed and later a penis constructed. Joan became John again and married a woman.
Evidence is accumulating that gender identification is determined during the development of the fetus due to genetics and/or hormonal environment. Female rodents exposed to androgens in utero exhibit male mating behavior. Females with congenital adrenal hyperplasia have been exposed to excess androgens in utero also engage in more masculine behavior such as "rough and tumble" play and are sexually attracted to females. Animal models of gonadal hormone influences on sexual differentiation of brain and behavior have been studied and also several clinical intersex cases including patients with congenital adrenal hyperplasia, a range of androgen insensitivities, 5 alpha reductase deficiency, and those whose mothers had hormones administered during pregnancy. Conclusions are that the prenatal or neonatal hormone environment contributes to the development of human behaviors that show sex differences, particularly childhood play behavior, sexual orientation and gender identity. There is also some evidence for their influence on aggression and cognition.



A gene was mapped to Xq28 for homosexuality in the male. It has been called GAY. You will find a discussion of this and other studies in OMIM entry #306995. LeVay measured the volume of four cell groups in the anterior hypothalamus of the post mortem brains of women, heterosexual males and homosexual males. He found that the interstitial nucleus of heterosexual males was more than twice as large than in women or homosexual males. The validity of this finding has been challenged by a number of scientists. This and other genetic research into male homosexuality has focused solely on the X chromosome, passed down to boys by their mother. A study by KIrsch (2005) looked at autosomes (non-sex chromosomes) of 456 individuals from 146 families with two or more gay brothers. It was found that among gay siblings, there were several identical stretches of DNA were shared among gay siblings on chromosomes 7, 8, and 10. About 60% of these brothers shared identical DNA on the three chromosomes where only 50% was expected if it were only due to chance. The region found on chromosome 10 correlated with sexual orientation only when it was inherited from the mother. This work will have to be confirmed by further studies and additional studies done to identify the genes within the newly discovered sequences that may be linked to sexual orientation. While some studies have shown that genes may play an important role in determining homosexuality most believe that other factors are also important in sexual orientation.
